{kind=link}
Citation:
Date Published:
27 January 2016Abstract:
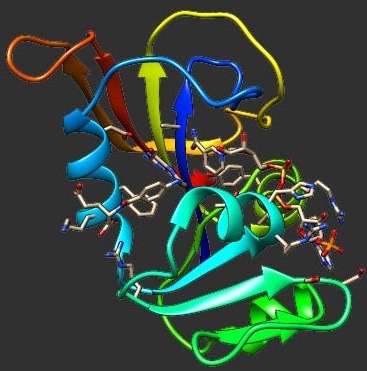
Fitness landscapes of drug resistance constitute powerful tools to elucidate mutational pathways of antibiotic escape. Here, we developed a predictive biophysics-based fitness landscape of trimethoprim (TMP) resistance for Escherichia coli dihydrofolate reductase (DHFR). We investigated the activity, binding, folding stability, and intracellular abundance for a complete set of combinatorial DHFR mutants made out of three key resistance mutations and extended this analysis to DHFR originated from Chlamydia muridarum and Listeria grayi. We found that the acquisition of TMP resistance via decreased drug affinity is limited by a trade-off in catalytic efficiency. Protein stability is concurrently affected by the resistant mutants, which precludes a precise description of fitness from a single molecular trait. Application of the kinetic flux theory provided an accurate model to predict resistance phenotypes (IC50) quantitatively from a unique combination of the in vitro protein molecular properties. Further, we found that a controlled modulation of the GroEL/ES chaperonins and Lon protease levels affects the intracellular steady-state concentration of DHFR in a mutation-specific manner, whereas IC50 is changed proportionally, as indeed predicted by the model. This unveils a molecular rationale for the pleiotropic role of the protein quality control machinery on the evolution of antibiotic resistance, which, as we illustrate here, may drastically confound the evolutionary outcome. These results provide a comprehensive quantitative genotype–phenotype map for the essential enzyme that serves as an important target of antibiotic and anticancer therapies.