Abstract:
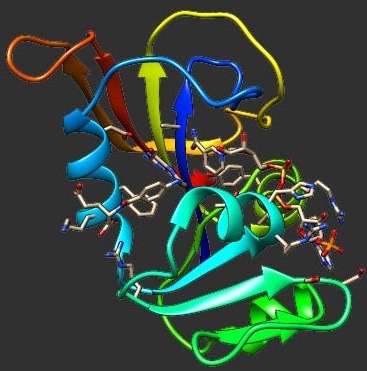
The BACE-1 enzyme is a prime target to find a cure to Alzheimer's disease. In this article, we used the MM-PBSA approach to compute the binding free energies of 46 reported ligands to this enzyme. After showing that the most probable protonation state of the catalytic dyad is mono-protonated (on ASP32), we performed a thorough analysis of the parameters influencing the sampling of the conformational space (in total, more than 35 μs of simulations were performed). We show that ten simulations of 2 ns gives better results than one of 50 ns. We also investigated the influence of the protein force field, the water model, the periodic boundary conditions artifacts (box size), as well as the ionic strength. Amber03 with TIP3P, a minimal distance of 1.0 nm between the protein and the box edges and a ionic strength of
I = 0.2 M provides the optimal correlation with experiments. Overall, when using these parameters, a Pearson correlation coefficient of
R = 0.84 (
R2 = 0.71) is obtained for the 46 ligands, spanning eight orders of magnitude of
Kd (from 0.017 nm to 2000 μM, i.e., from −14.7 to −3.7 kcal/mol), with a ligand size from 22 to 136 atoms (from 138 to 937 g/mol). After a two-parameter fit of the binding affinities for 12 of the ligands, an error of RMSD = 1.7 kcal/mol was obtained for the remaining ligands.
Publisher's Version
{kind=link}