Abstract:
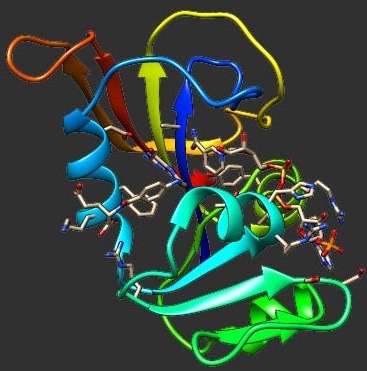
Recent experiments and simulations have demonstrated that proteins can fold on the ribosome. However, the extent and generality of fitness effects resulting from cotranslational folding remain open questions. Here we report a genome-wide analysis that uncovers evidence of evolutionary selection for cotranslational folding. We describe a robust statistical approach to identify loci within genes that are both significantly enriched in slowly translated codons and evolutionarily conserved. Surprisingly, we find that domain boundaries can explain only a small fraction of these conserved loci. Instead, we propose that regions enriched in slowly translated codons are associated with cotranslational folding intermediates, which may be smaller than a single domain. We show that the intermediates predicted by a native-centric model of cotranslational folding account for the majority of these loci across more than 500
Escherichia coli proteins. By making a direct connection to protein folding, this analysis provides strong evidence that many synonymous substitutions have been selected to optimize translation rates at specific locations within genes. More generally, our results indicate that kinetics, and not just thermodynamics, can significantly alter the efficiency of self-assembly in a biological context.
Publisher's Version
{kind=link}