Date Published:
2001
Abstract:
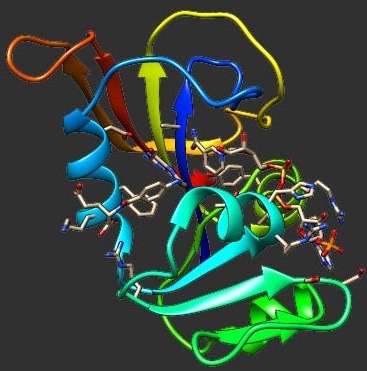
We present a novel Monte Carlo simulation of protein folding, in which all heavy atoms are represented as interacting hard spheres. This model includes all degrees of freedom relevant to folding, all side-chain and backbone torsions, and uses a Gō potential. In this study, we focus on the 46 residue α/β protein crambin and two of its structural components, the helix and helix hairpin. For a wide range of temperatures, we recorded multiple folding events of these three structures from random coils to native conformations that differ by less than 1 Å Cα dRMS from their crystal structure coordinates. The thermodynamics and kinetic mechanism of the helix-coil transition obtained from our simulation shows excellent agreement with currently available experimental and molecular dynamics data. Based on insights obtained from folding its smaller structural components, a possible folding mechanism for crambin is proposed. We observed that the folding occurs via a cooperative, first order-like process, and that many folding pathways to the native state exist. One particular sequence of events constitutes a "fast-folding” pathway where kinetic traps are avoided. At very low temperatures, a kinetic trap arising from the incorrect packing of side-chains was observed. These results demonstrate that folding to the native state can be observed in a reasonable amount of time on desktop computers even when an all-atom representation is used, provided the energetics sufficiently stabilize the native state.
Website
{kind=link}