Date Published:
2012
Abstract:
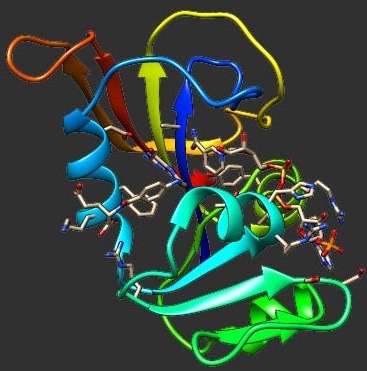
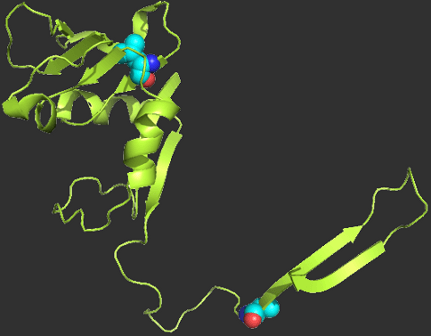
Gō models are exceedingly popular tools in computer simulations of protein folding. These models are native-centric, i.e., they are directly constructed from the protein's native structure. Therefore, it is important to understand up to which extent the atomistic details of the native structure dictate the folding behavior exhibited by Gō models. Here we address this challenge by performing exhaustive discrete molecular dynamics simulations of a Gō potential combined with a full atomistic protein representation. In particular, we investigate the robustness of this particular type of Gō models in predicting the existence of intermediate states in protein folding. We focus on the N47G mutational form of the Spc-SH3 folding domain (x-ray structure) and compare its folding pathway with that of alternative native structures produced in silico. Our methodological strategy comprises equilibrium folding simulations, structural clustering, and principal component analysis.
Website
{kind=link}